MADC File Format
Description of the DArT MADC file
Explanation of the MADC file format and the conversion to fixed allele ID at BI.
flowchart TD
A(["Genotype Sequencing"]) --> B["MADC File"]
B --> D["Fixed Allele IDs Assigned"]
D --> n3["Kinship Matrix"] & n4["Convert to VCF"]
B@{ shape: div-proc}
D@{ shape: procs}
n3@{ shape: db}
n4@{ shape: db}
A:::Aqua
B:::Sky
D:::Sky
n3:::Peach
n4:::Peach
classDef Aqua stroke-width:1px, stroke-dasharray:none, stroke:#46EDC8, fill:#DEFFF8, color:#378E7A
classDef Sky stroke-width:1px, stroke-dasharray:none, stroke:#374D7C, fill:#E2EBFF, color:#374D7C
classDef Peach stroke-width:1px, stroke-dasharray:none, stroke:#FBB35A, fill:#FFEFDB, color:#8F632D
DArT MADC File
The DArT MADC (Missing Allele Discovery Counts) file is a comma-separated file provided by DArT from a sequencing project. It includes the following key columns:
- AlleleID: Typically contains the chromosome, position, reference or alternative allele designation, and the allele ID according to the haplotype database (e.g.,
Chr01_000084128|Ref_0001). - CloneID: Represents the chromosome and position (e.g.,
Chr01_000084128). - AlleleSequence: The amplicon sequence (e.g.,
GAGTGTGAAGATTTGGACAAAAGAGGTTGGTTTTTACTGTTATGGCATTTATCTCCTTATAAAATTTTGTATTTTTTTTGT). - Sample Columns: Named with sample IDs and containing counts of each allele per sample.
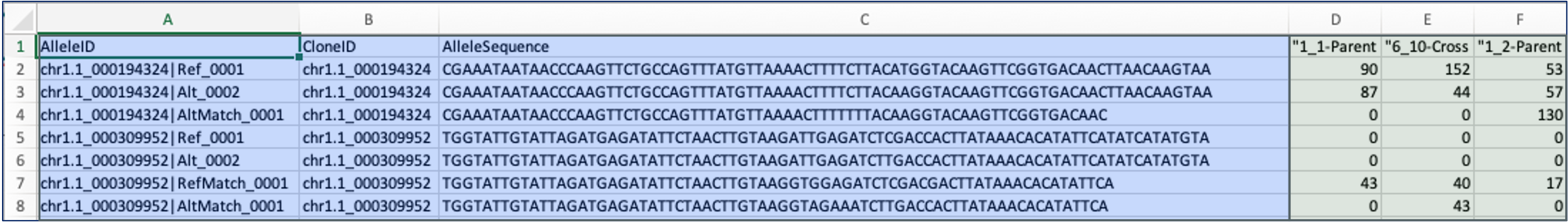 Example Missing Allele Discovery (MADC) file with fixed allele IDs
Example Missing Allele Discovery (MADC) file with fixed allele IDs
Important: BIGr requires fixed AlleleIDs tagged with a respective number (e.g., suffix: _0001). Fixed AlleleIDs are generated by matching the MADC contents to the haplotype database. If your MADC file does not contain fixed AlleleIDs, please contact Breeding Insight.
Target vs. Off-Target SNPs
If you provide a MADC file, you can choose between extracting:
- Only target SNPs – These are the SNPs for which probes were specifically designed. If your panel includes 3,000 SNPs, your output VCF file should contain exactly 3,000 target SNPs.
- Both target and off-target SNPs – In addition to target SNPs, BIGr will identify other SNPs within the amplicon region.
Target SNPs
If you decide to extract only the target SNPs, you can choose if reference (REF) and alternative (ALT) bases should also be extracted. For extracting this information from the MADC file, BIGr compares the reference sequences with the alternative, finds the polymorphic site and notate the changing base. This requires:
- Reference (
Ref_0001) and alternative (Alt_0002) sequences for each tag. If one or both is missing, the tag will be discarded. - A single polymorphism between them. If more than one is found, the tag is discarded
- A
.botlocifile to inform which tag sequences should be converted to its reverse complement. For BI-supported marker panel species this file is already embedded within the app, no upload is required.
If you choose to not extract REF and ALT information, all tags will be kept, but your VCF will have missing data (.) in the REF and ALT fields. The only process in BIGr that will require this information is the Dosage Calling with PolyRAD.
Target and off-target (all) SNPs
To identify off-target SNPs, BIGr aligns each amplicon against its reference (via Bioconductor’s Biostrings + pwalign) to uncover additional polymorphisms. This procedure requires:
- Reference & alternative sequences (e.g.
Ref_0001,Alt_0002) for every tag- If a FASTA haplotype database is supplied, any missing sequences in the MADC file will be retrieved automatically.
- Without a FASTA database, tags lacking either sequence are discarded.
- Exactly one polymorphic site
- Tags with zero or multiple variants are excluded.
.botlocifile- Specifies which tags must be reverse-complemented. For BI-supported species, this file is bundled within the app—no upload needed.